W ciągu ostatnich miesięcy na portalach społecznościowych oraz polskich i zagranicznych serwisach związanych ze środo wiskiem chorych na mukowiscydozę pojawiło się wiele, często sensacyjnych informacji na temat nowych możliwości leczenia choroby. Również w gabinecie rodzice pytają o nowe leki, podekscytowani informacjami ze świata i pełni nadziei na przełom w leczeniu. W tym artykule postaram się wyjaśnić, ile obecnie wiemy i jakie powinny być realne oczekiwania w stosunku do leków z grupy potencjatorów i korektorów białka CFTR.
Główny winowajca choroby – białko CFTR
Przyczyną mukowiscydozy jest mutacja w genie, który koduje białko tworzące kanał jonowy określany skrótem CFTR. (ang. cystic fibrosis transmembrane conductance regulator). Droga od mutacji do choroby jest już dobrze poznana i obejmuje kaskadę zdarzeń nieuchronnie prowadzących do zniszczenia płuc i innych ważnych narządów (tab. 1).
Tabela 1. Patogeneza mukowiscydozy – sekwencja zdarzeń (oprac. własne)
| Mutacja w genie CFTR Brak lub nieprawidłowe działanie kanału CFTR Nieprawidłowy przepływ NaCl i wody przez błonę komórkową Odwodnienie i zwiększenie lepkości śluzu Upośledzony ruch rzęsek nabłonka oddechowego Zaleganie wydzieliny i obturacja dróg oddechowych Przewlekły stan zapalny i kolonizacja bakteryjna Destrukcja tkanki płucnej Utrata funkcji płuc prowadząca ostatecznie do niewydolności oddechowej |
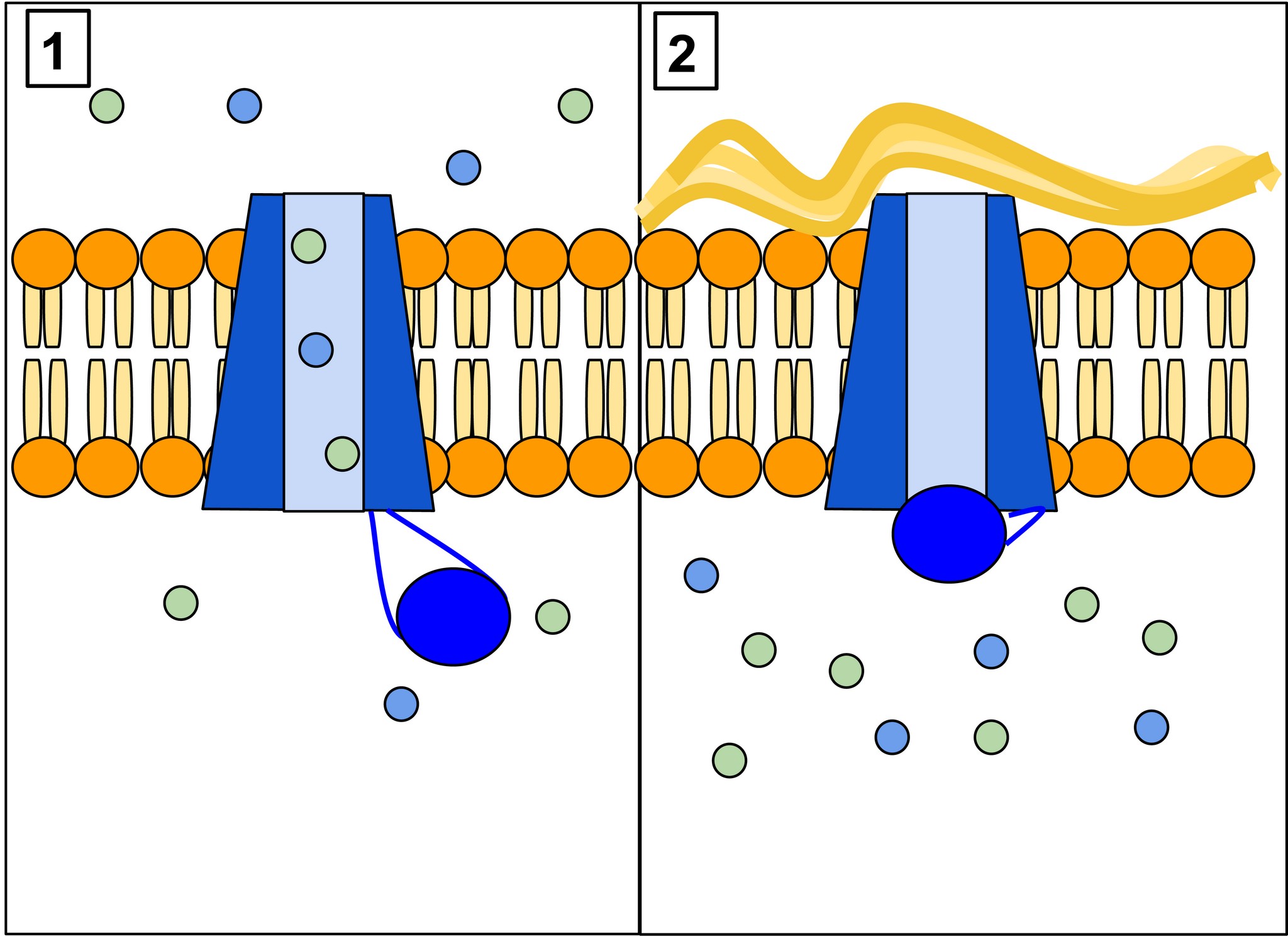
Białko CFTR (a właściwie glikoproteina) jest jednym z największych białek ludzkiego organizmu, składa się 1480 aminokwasów, a informacja genetyczna zapisana jest w 230 tys. par zasad ludzkiego DNA. Występuje ono w komórkach nabłonkowych wielu narządów takich jak płuca, wątroba, trzustka, jelita czy skóra. Powstawanie tej dużej cząsteczki jest skomplikowane, rozpoczyna się w jądrze komórkowym, gdzie materiał genetyczny przepisywany jest na łańcuch mRNA w postaci długiej nici, następnie trafia do cytoplazmy, gdzie na podstawie mRNA powstaje łańcuch aminokwasów, przypominający wagony długiego pociągu. Następnie nić formuje trójwymiarową strukturę, która ostatecznie trafia do błony komórkowej, gdzie tworzy rodzaj bramki – kanału dla jonów, głównie chlorkowych (Cl–). U osób zdrowych kanał ten przepuszcza jony Cl– na zewnątrz komórki, za nimi podąża sód (Na+) oraz woda, tworząc płynną warstwę otaczającą powierzchnię komórek nabłonkowych tworzących pofałdowania zwane mikrokosmkami (rysunek 1–3). Ruch mikrokosmków zapewnia barierę ochroną dla komórek i umożliwia usuwanie wydzieliny, na przykład z dróg oddechowych. U osób chorych kanał CFTR nie działa prawidłowo lub w ogóle nie jest wytwarzany, czego konsekwencją jest obecność gęstszej (odwodnionej) wydzieliny uszkadzającej komórki nabłonka.
Mutacja mutacji nierówna
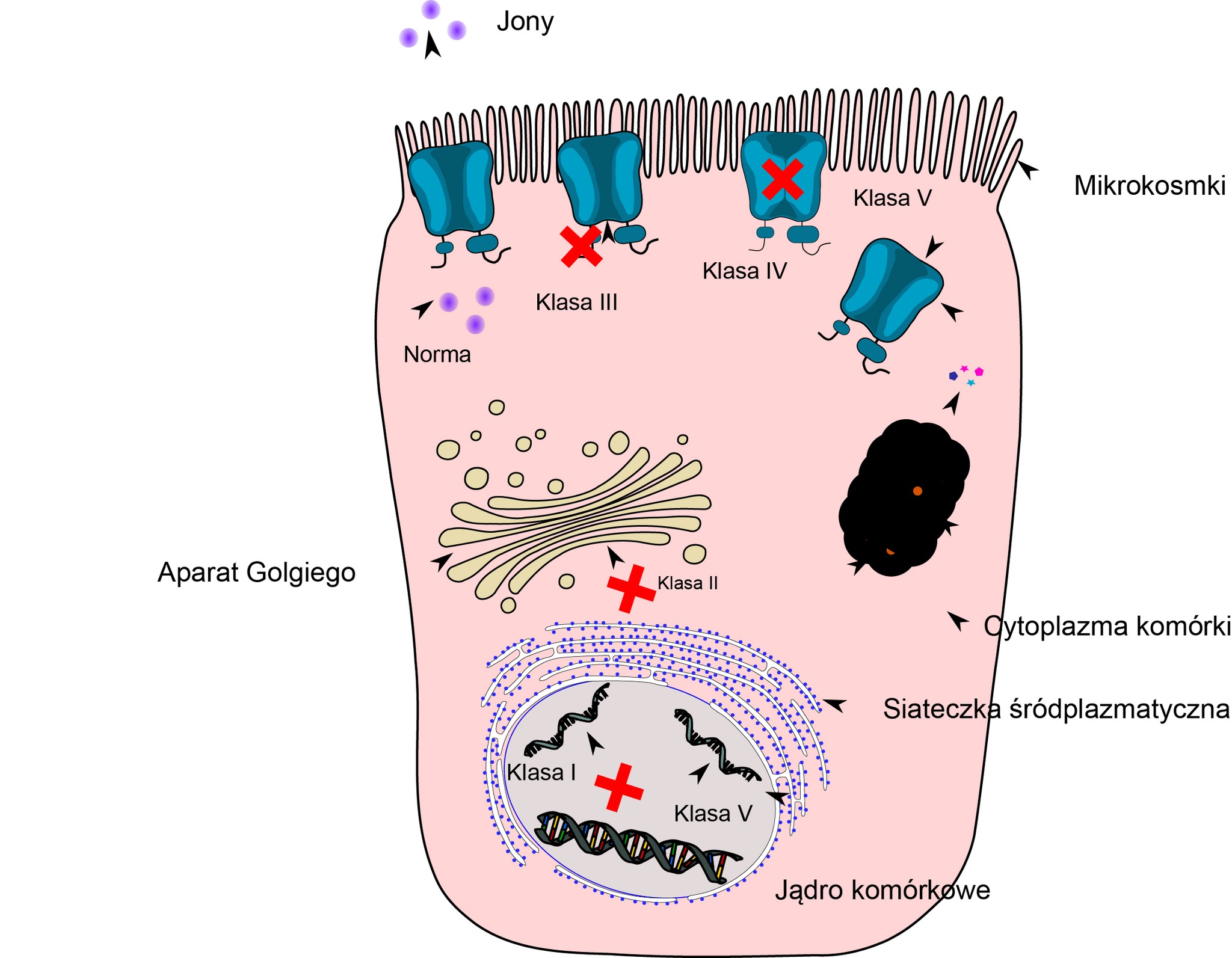
Jak wcześniej wspomniano, skutkiem mutacji w materiale genetycznym (DNA) jest wada kanału jonowego. Ta wada może mieć różny charakter zarówno jakościowy, jak i ilościowy, to znaczy mutacja może powodować całkowite zatrzymanie produkcji białka (czyli jego zupełny brak w błonie komórkowej), jego zniszczenie w cytoplazmie lub tylko nieznaczne upośledzenie w działaniu, objawiające się na przykład gorszym przewodzeniem jonów Cl–. Te różnice w mechanizmie powstawania wady kanału CFTR stały się podstawą do podziału wszystkich mutacji na 5 klas (tab. 2).
Tabela 2. Klasyfikacja mutacji genu CFTR (Hodson and Goddes’ Cystic Fibrosis, 4ed, 2016)
| Klasa | Wpływ na CFTR | Działanie CFTR | Obecność CFTR w błonie komórki | Przykład | Możliwość leczenia |
|---|---|---|---|---|---|
| I | Błąd na drodze syntezy białka CFTR (mutacja nonsens) | Nie | Nie | G542X, W1282X, R553X | Przepuszczenie kodonu non sensownego (ataluren), inhibitor NMD |
| II | błąd na drodze obróbki białka CFTR | Nie | Nie | F508del, N1303K, I507del | Korektor (lumacaftor) |
| III | nieprawidłowa regulacja lub wadliwe bramkowanie kanału CFTR | Nie | Tak | G178R, G551D, G551S, R560T | Potencjator (ivacaftor) |
| IV | nieprawidłowe przewodzenie jonów chlorkowych przez kanał CFTR | Zmniejszona | Tak | R117H, R334W, R347P, R1070W | Potencjator (ivacaftor) |
| V | zmniejszona ilość kanałów CFTR | Zmniejszona | zmniejszona | 3272-26A>G, 3849+10kbC>T, A455E, D565G 3120+1G>A | inhibitor NMD, modulatory splicingu |
Ma to obecnie również przełożenie praktyczne – istnienie różnych mutacji pozwala odpowiedzieć, dlaczego chorzy na mukowiscydozę mogą mieć odmienny przebieg choroby, od pełnoobjawowej z typową niewydolnością trzustki i szybkim pogorszeniem objawów płucnych, do bardzo łagodnej z pojedynczymi objawami, takimi jak nawrotowe zapalenie zatok czy niepłodność. Sprawę komplikuje jednak fakt, że pacjenci nawet z tą samą mutacją różnie chorują, co dowodzi istnienia innych czynników modyfikujących działanie białka CFTR i jest przedmiotem intensywnych badań. Ogólnie mówiąc, można przyjąć, że mutacje należące do I-III klasy najczęściej skutkują pełnoobjawowym obrazem choroby, mutacje klasy IV i V są klinicznie łagodniejsze, z zastrzeżeniem, że dość często obserwuje się zmienność kliniczną.
Trochę statystyki
Obecnie znanych jest 2007 mutacji genu CFTR (baza danych CFMDB, marzec 2016), jednak tylko około 20 występuje z częstością większą niż 0,1%, a jedynie 150 jest uznawane z pewnością za wywołujące mukowiscydozę (tabela 3 i 4). 1 2Są to najczęściej mutacje punktowe typu missens albo o charakterze małych insercji, rzadziej mutacje typu nonsens i na złączach intronekson. Tylko wyjątkowo są to duże delecje.
Tabela 3. Częstość mutacji na świecie (Hodson and Goddes’ Cystic Fibrosis, 4ed, 2016)
| Rodzaj mutacji | Częstość |
|---|---|
| delF508 | 66,0% |
| G542X | 2,4% |
| G551D | 1,6% |
| N1303K | 1,3% |
| W1282X | 1,2% |
| R553X | 0,7% |
| 621+1G>T | 0,7% |
| 1717-1G>A | 0,6% |
| R117H | 0,3% |
Tabela 4. Częstość mutacji z uwzględnieniem rodzaju (Wikipedia)
| Rodzaj mutacji | Liczba | Częstość |
|---|---|---|
| Typu missens (zmiana kodonu) | 795 | 39,61 |
| Przesunięcie ramki odczytu | 313 | 15,60 |
| Błędy na poziomie splicingu | 228 | 11,36 |
| Typu nonsense | 167 | 8,32 |
| Insercje lub delecje w obrębie ramki odczytu | 40 | 1,99 |
| Duże insercje lub delecje | 52 | 2,59 |
| Mutacje promotora genu | 15 | 0,75 |
| Zmienność sekwencji | 269 | 13,40 |
| Nieznane | 128 | 6,38 |
W Polsce najczęstsza mutacja odpowiada za około 70% przypadków choroby, oznaczana jest jako F508del i wiąże się z ciężkim przebiegiem choroby. Mutacja ta polega na delecji trzech nukleotydów, co powoduje usunięcie fenyloalaniny z sekwencji aminokwasowej białka. Tak zmienione białko jest rozpoznawane przez mechanizmy naprawcze komórki i degradowane (klasa II mutacji). W przypadku mutacji W1282X dochodzi do przedwczesnego zakończenia procesu transkrypcji i znacznego skrócenia białka CFTR. Ta mutacja jest szczególnie częsta wśród Żydów aszkenazyjskich i odpowiada za prawie 40% przypadków choroby. Innym przykładem nieprawidłowości jest mutacja G551D występująca u około 5% chorych i powodująca całkowicie zamknięcie kanału chlorkowego.
Inne mutacje, na przykład R117H, R334W i R347P, odpowiadają za łagodniejszy przebieg choroby. W Europie Wschodniej i Środkowej, także w Polsce, spotykana jest mutacja CFTRdele2,3 (duża delecja 21 800 par zasad), prawie w ogóle nie spotykana w innych populacjach 3. Mutacja ta również wywołuje ciężką postać choroby, z niewydolnością trzustki i wczesnym początkiem.
Podziałajmy w komórce
Dotychczas, poza pojedynczymi próbami terapii genowej, standardowe leczenie skupiało się na leczeniu skutków choroby i obejmowało antybiotykoterapię, leczenie żywieniowe, fizykoterapię, aerozoloterapię, przyjmowanie leków mukolitycznych, tlenoterapię domową oraz w ostateczności transplantację płuc. Jednak pod koniec ubiegłej dekady pojawiła się zupełnie nowa koncepcja leczenia, która miała w efekcie poprawić funkcjonowanie kanału jonowego CFTR, czyli istotę choroby. Terapia ta polega na podaniu choremu substancji, która zmodyfikuje kanał jnowy tam, gdzie pojawia się problem. Jeśli łańcuch białkowy ulega zbyt wczesnemu zakończeniu (mutacja nonsensowna), lek blokuje informację stop i synteza trwa dalej. Jeśli kanał CFTR nie przewodzi jonów Cl– w sposób prawidłowy, cząsteczka leku go modyfikuje i zapewnia lepsze działanie. Leczenie to nie ma na celu wyleczenia mukowiscydozy, jednak jego efekt, przynajmniej teoretycznie, powinien być znacznie lepszy niż jakiekolwiek dotychczas stosowane leczenie.
Ivacaftor – poznajmy się bliżej
Jak do tej pory, jednym z pierwszych leków działających na poziomie komórki jest ivacaftor, oznaczany pierwotnie jako VX-770, a obecnie znany pod nazwą handlową Kalydeco. Ivacaftor jest tak zwanym potencjatorem kanału CFTR, to znaczy zwiększa prawdopodobieństwo otwarcia kanału w mutacjach klasy III (m.in. G551), które występują u kilku procent chorych. Badania przedkliniczne leku pokazały poprawę uwodnienia i ruchomości rzęsek nabłonka u chorych na mukowiscydozę, a badania II fazy poprawę funkcjonowania kanałów CFTR w nabłonku nosa i gruczołów potowych. Zauważalna była również tendencja do normalizacji stężenia chlorków w pocie poniżej 60 mmol/L 4. W randomizowanym badaniu STRIVE obejmującym dorosłych oraz dzieci z FEV1 40–105% (parametr czynności płuc mierzony za pomocą spirometrii) w 24 tygodniu leczenia wykazano istotną poprawę tego parametru o 10,6% 5. Efekt leku był widoczny już po 15 dniach i trwał przez okres trwania badania wynoszącego 48 tygodni. U blisko 75% dorosłych pacjentów obserwowano poprawę funkcji płuc 5%, a poprawa była widoczna także u pacjentów z zaawansowaną chorobą. Zmniejszyło się również ryzyko zaostrzeń oskrzelowo-płucnych o 55% oraz stężenie chlorków w teście potowym aż o 48 mmol/L. Efekt ten również utrzymywał się przez cały czas badania. W innym badaniu poprawę wykazano również u dzieci przedszkolnych z prawidłowymi badaniami czynnościowymi (wzrost FEV1 o 7% w 28. dniu badania) 6 Obecnie toczy się kilka badań klinicznych, także w innych mutacjach klasy III.
Łyżka dziegciu w beczce miodu
Badania kliniczne ivacaftoru z pewnością są bardzo obiecujące, jednak należy pamiętać o dwóch rzeczach. Po pierwsze wciąż mało wiemy na temat skuteczności leku. Poprawa pewnych parametrów, takich jak FEV1, liczba zaostrzeń oskrzelowo-płucnych czy stan odżywienia jest istotna, jednak najważniejsza jest długookresowa ocena kliniczna oraz tak zwane twarde punkty końcowe (przeżywalność, jakość życia, czas i liczba hospitalizacji), a na to potrzeba kilku lat wnikliwych badań. Druga sprawa to koszt leku. Jak wynika z informacji prasowych, roczna terapia Kalydeco to kwota 300 tys. dolarów, co wydaje się sumą zawrotną, szczególnie w polskich realiach. W czerwcu 2015 r. Agencja Oceny Technologii Medycznej negatywnie zaopiniowała refundację leku Kalydeco we wskazaniu: leczenie mukowiscydozy u pacjentów w wieku 6 lat i starszych, z jedną z następujących mutacji bramkowania genu CFTR (klasy III): G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N lub S549R. W uzasadnieniu stanowiska można przeczytać:„Inkrementalny współczynnik kosztów użyteczności (ICUR) dla stosowania produktu Kalydeco wielokrotnie przekracza próg przyjęty w Polsce dla technologii efektywnych kosztowo. Rozpatrywany lek jest finansowany w zaledwie 1 kraju (Grecji) o zbliżonym do Polski poziomie PKB per capita, w pozostałych 6 nie jest finansowany ze środków publicznych”. Z powyższego można wnioskować, że przynajmniej na jakiś czas droga ivacaftoru do wpisania na listę refundacyjną została zamknięta.
Inni gracze
VX-809 to symbol leku o nazwie lumacaftor, którego celem są mutacje klasy II, m.in. najczęściej występującej w Polsce F508del. Ułatwia on „dojrzewanie” trójwymiarowej struktury białka CFTR i zwiększa aktywność kanałów do 15%8, 9. 7 8 Ponieważ lek modyfikuje cząsteczkę kanału jonowego, nazywany jest korektorem. Wstępne badania pokazały, że poprawa funkcji CFTR jest zauważalna w nabłonku gruczołów potowych, natomiast nie zaobserwowano zmiany w nabłonku nosa i odbytnicy.
Kilka kolejnych badań przyniosło rozczarowanie skutecznością lumacaftoru, producent leku (Vertex Pharmaceuticals) zdecydował się na przeprowadzenie badań nad jednoczesnym zastosowaniem potencjatora (ivacaftor) i korektora (lumacaftor). W ten sposób powstał lek o nazwie Orkambi.
W międzynarodowym badaniu II fazy wykazano istotny statystycznie spadek stężenia Cl– w teście potowym o 9 mmol/L, a także wzrost wartości FEV1 o 5,6% 9. W lipcu 2015 opublikowano wyniki 2 randomizowanych badań III fazy TRAFFIC i TRANSPORT, w których wzięli udział pacjenci z 2 mutacjami F508del powyżej 12. roku życia 10. Po 24 tygodniach leczenia średnia poprawa FEV1 w grupie leczonej wynosiła 2,6–4%, a średni spadek częstości zaostrzeń wyniósł 30–39% w stosunku do grupy placebo. Kolejne badania są w toku. Na razie trudno wyrokować co do skuteczności leku w populacji dziecięcej, tym bardziej, że jest to dość niejednorodna grupa.
Kolejnym lekiem, którego celem są mutacje klasy I jest Ataluren (PTC124). Lek ten wymusza kontynuowanie syntezy CFTR mimo obecności kodonu nonsensownego, powodując pojawienie się kanałów jonowych na powierzchni błony komórkowej. Kliniczne badania II fazy dają sprzeczne wyniki. Badania francuskie i izraelskie pokazały przywrócenie funkcji CFTR, jednak badania amerykańskie tego nie potwierdziły. 11 12 13Również w randomizowanym badaniu III fazy nie wykazano istotnej poprawy funkcji płuc w populacji pacjentów z mutacjami nonsensownymi 14.
Co dalej?
Obecnie jesteśmy świadkami rewolucji w leczeniu mukowiscydozy – zaczynamy oddziaływać na chorobę na poziomie komórkowym, a nie skupiać się jedynie na jej skutkach. Już dziś wiadomo, że nowe leki są skuteczne, a w przyszłości będą jeszcze bardziej, na przykład przy połączeniu 2 substancji. Niezmiennie podstawowym problemem będą środki finansowe. Liczba kosztownych terapii z roku na rok wzrasta (nowotwory, terapia biologiczna, zakażenie WZW typu C, choroby rzadkie), a nakłady na ochronę zdrowia rosną tylko nieznacznie. Ta dysproporcja między możliwościami medycyny a możliwościami finansowania systemu ochrony zdrowia są widoczne we wszystkich rozwiniętych krajach. Kosztowne terapie to także problem etyczny, komu zapewnić leczenie, jakiej skuteczności należy oczekiwać, przez jaki czas finansować terapię, kto ma pierwszeństwo. W przestrzeni publicznej brakuje takiej debaty.
W obecnej chwili publiczne finansowanie potencjatorów i korektorów kanału CFTR wydaje się być nierealne. Natomiast warto skupić się na terapiach, która są w zasięgu ręki, a które są równie skuteczne. Mowa tu na przykład o dostępie do wziewnej tobramycyny, która ma przewagę nad wziewną kolistyną, a która jest dostępna jedynie dla wybranych pacjentów w ramach programu lekowego NFZ.
Bibliografia
- Araujo F.G. et al., Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil. Braz J Med Biol Res, 2005, 38 (1): s. 11–15.
- Boyle M.P. et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med, 2014, 2 (7): s. 527–538.
- Clancy J.P. et al. No detectable improvements in cystic fibrosis transmembrane conductance regulator by nasal aminoglycosides in patients with cystic fibrosis with stop mutations. Am J Respir Cell Mol Biol, 2007, 37 (1): s. 57–66.
- Dork T. et al., Characterization of a novel 21-kb deletion, CFTRdele2,3(21 kb), in the CFTR gene: a cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum Genet, 2000, 106 (3): s. 259–268.
- http://www.genet.sickkids.on.ca
- Kerem E. et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, doubleblind, placebo-controlled phase 3 trial. Lancet Respir Med, 2014, 2 (7): s. 539–547.
- Kerem E. et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet, 2008, 372 (9640): s. 719–727.
- McKone E.F. et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-<em>CFTR</em> mutation: a phase 3, open-label extension study (PERSIST). The Lancet Respiratory Medicine. 2 (11): s. 902–910.
- Ramsey B.W. et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med, 2011, 365 (18): s. 1663–1672.
- Quittner A. et al. Effect of ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation: patient-reported outcomes in the STRIVE randomized, controlled trial. Health Qual Life Outcomes, 2015, 13: s. 93.
- Sermet-Gaudelus I. et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med, 2010, 182 (10): s. 1262–1272.
- Van Goor F. et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A, 2011, 108 (46): s. 18843–18848.
- Van Goor F. et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol, 2006, 290 (6): s. L1117–1130.
- Wainwright C.E., et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med, 2015, 373 (3): s. 220–231.
Przypisy
- Araujo F.G., et al., Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil. Braz J Med Biol Res, 2005, 38 (1): s. 11–15. ↩︎
- http://www.genet.sickkids.on.ca. ↩︎
- Dork T., et al., Characterization of a novel 21-kb deletion, CFTRdele2,3(21 kb), in the CFTR gene: a cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum Genet, 2000, 106 (3): s. 259–268. ↩︎
- Quittner A., et al. Effect of ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation: patientreported outcomes in the STRIVE randomized, controlled trial. Health Qual Life Outcomes, 2015, 13: s. 93. ↩︎
- Ramsey B.W., et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med, 2011, 365 (18): s. 1663–1672. ↩︎
- McKone E.F., et al. Longterm safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-<em>CFTR</em> mutation: a phase 3, open-label extension study (PERSIST). The Lancet Respiratory Medicine. 2 (11): s. 902–910. ↩︎
- Van Goor F., et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A, 2011, 108 (46): s. 18843–18848. ↩︎
- Van Goor F., et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol, 2006, 290 (6): s. L1117–1130. ↩︎
- Boyle M.P., et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med, 2014, 2 (7): s. 527–538. ↩︎
- Wainwright C.E., et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med, 2015, 373 (3): s. 220–231. ↩︎
- Clancy J.P., et al. No detectable improvements in cystic fibrosis transmembrane conductance regulator by nasal aminoglycosides in patients with cystic fibrosis with stop mutations. Am J Respir Cell Mol Biol, 2007, 37 (1): s. 57–66. ↩︎
- Kerem E., et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet, 2008, 372 (9640): s. 719–727. ↩︎
- Sermet-Gaudelus I., et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med, 2010, 182 (10): s. 1262–1272. ↩︎
- Kerem E., et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, doubleblind, placebocontrolled phase 3 trial. Lancet Respir Med, 2014, 2 (7): s. 539–547. ↩︎